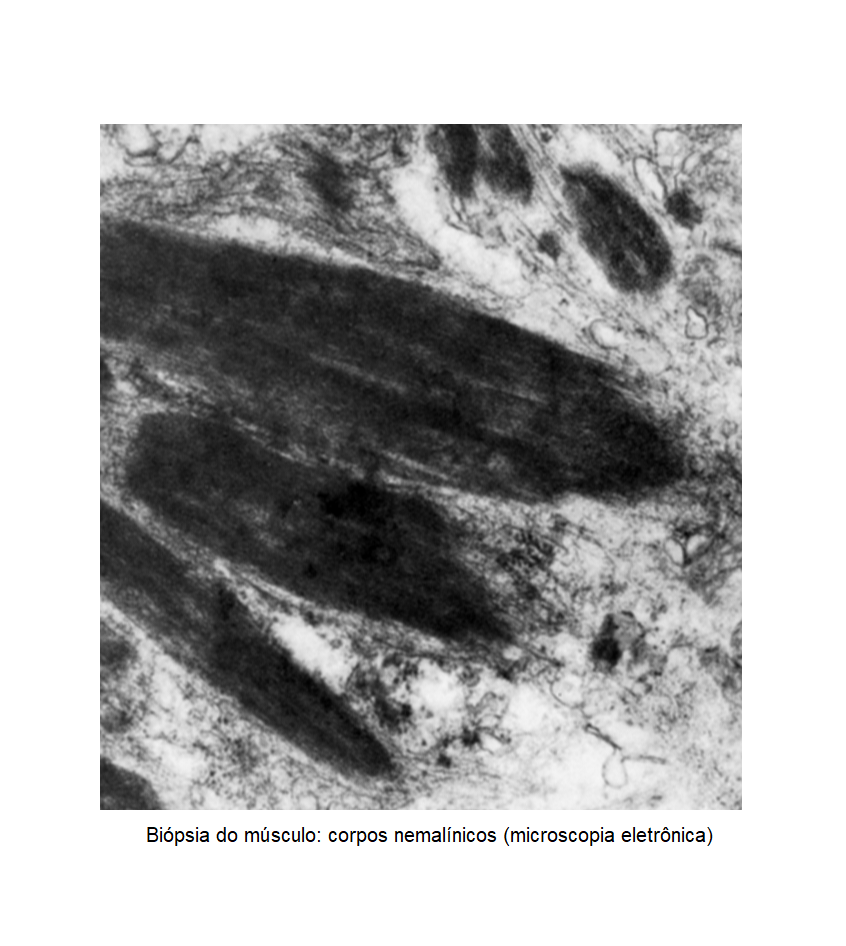
Introdução: A miopatia de Nemaline refere-se a um grupo de desordens neuromusculares de etiologia genética, que se caracteriza por leva à fraqueza muscular, resultante do acúmulo de corpúsculos bastonetiformes nas fibras musculares. Objetivo:Descrever um caso raro e diagnóstico diferencial de amiloidose. Relato: Paciente com quadro de dificuldade progressiva de levantar e subir escadas com piora lenta, evoluiu com intolerância ao exercício, fadiga, palpitações e dispneia aos mínimos esforços aos 35 anos. Ao início do acompanhamento com cardiologista foi identificado doença estrutural compatível com insuficiência cardíaca de fração reduzida (37%) e discreta hipertrofia ventricular concêntrica, com aumento biatrial. Na investigação, foi lançada como primeira hipótese amiloidose, então realizou eletroforese e imunofixação de proteínas kappa e lambda em sangue e urina (negativas), cintilografia com tecnécio (inconclusiva) e biópsia endomiocárdica com vermelho congo positivo porém sem hiperrefringência à luz polarizada. A eletroneuromiografia sugeria envolvimento muscular primário, realizada biópsia que revelou corpos nemalínicos clássicos e deficiência de fibras do tipo II. O teste genético demonstrou variante patogênica(p.Gly17Asp) de ACTA1 em heterozigose, confirmando o diagnóstico suspeito. Discussão: A miopatia de Nemaline cursa como principal manifestação a fraqueza de músculos proximais e sua expressão gênica envolve uma gama de sintomas, de leves a severos. Recém-nascidos com esta condição podem apresentar hipotonia e distúrbios gastroesofágicos importantes, já adultos apresentam um quadro mais tardio com comprometimento de marcha progressivo, alterações osteomusculares de cintura escapular e comprometimento miocárdico é variável, em sua maioria descrito como padrão restritivo que simula a cardiomiopatia amiloide. O diagnóstico costuma ser alcançado através de uma biópsia muscular com os corpúsculos bastonetiformes, porém não são patognomônicos, deve ser correlacionado com o quadro clínico, história familiar e teste genético. Até o momento, foram identificadas quatorze variações patogênicas em genes diferentes (ACTA1, TPM3, TPM2, CFL2, KBTBD13, KLHL40, KLH41, LMOD3, MYPN, TNNT1, SEPN1, RYR1, MYH7 e NEB), que confirmam a complexidade e heterogenicidade na avaliação do espectro clínico. Conclusão: As miopatias congênitas são doenças musculares genéticas caracterizadas por hipotonia e fraqueza muscular, sua forma de apresentação é variável e os diagnósticos diferenciais devem ser revisados, principalmente na era da medicina de precisão.